
-----BEGIN PGP SIGNED MESSAGE----- Hash: SHA1
You may have been patiently waiting for the first installment of my nanotechnology blog and you're right to expect great things. I have found myself pushing more and more stuff onto my stack and there will be a time for popping all those off the stack. The first one is what I've been working on the past three weeks.
There are plenty of tutorials for GROMACS but because I had a bunch of problems using it, I thought I would write another one. My tutorial is based on this one, so if you want the original, look there.
First, we need a molecule to minimize*. We could use 1OEI, but let's start with a blank slate. Minimizing an already minimized protein is kinda boring.
PyMOL allows you to create proteins from scratch. Use the builder to create a simple two-residue peptide, acetyl-alanine. You can do this by clicking Builder, then Protein, then Ace, then Create As New Object. Then simply click Ala. You can also do this with two lines of Python pasted directly into the command prompt:
editor.attach_amino_acid("pk1", "ace", object="obj1", _self=cmd)
editor.attach_amino_acid("pk1", "ala", _self=cmd)
Another way to do this is even easier:
editor.build_peptide('BA')
Either way, save your work using the save command.
save aceala1.pdbNow we're ready to use GROMACS.
pdb2gmx -f aceala1.pdb -o aceala1
:-) G R O M A C S (-:
Gnomes, ROck Monsters And Chili Sauce
:-) VERSION 4.6.5 (-:
Contributions from Mark Abraham, Emile Apol, Rossen Apostolov,
Herman J.C. Berendsen, Aldert van Buuren, Pär Bjelkmar,
Rudi van Drunen, Anton Feenstra, Gerrit Groenhof, Christoph Junghans,
Peter Kasson, Carsten Kutzner, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2012,2013, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.
:-) pdb2gmx (-:
Option Filename Type Description
- - ------------------------------------------------------------
-f aceala1.pdb Input Structure file: gro g96 pdb tpr etc.
-o aceala1.gmx.gro Output Structure file: gro g96 pdb etc.
-p topol.top Output Topology file
-i posre.itp Output Include file for topology
-n clean.ndx Output, Opt. Index file
-q clean.pdb Output, Opt. Structure file: gro g96 pdb etc.
Option Type Value Description
- - ------------------------------------------------------
- - -[no]h bool no Print help info and quit
- - -[no]version bool no Print version info and quit
- - -nice int 0 Set the nicelevel
- - -chainsep enum id_or_ter Condition in PDB files when a new chain should
be started (adding termini): id_or_ter,
id_and_ter, ter, id or interactive
- - -merge enum no Merge multiple chains into a single
[moleculetype]: no, all or interactive
- - -ff string select Force field, interactive by default. Use -h for
information.
- - -water enum select Water model to use: select, none, spc, spce,
tip3p, tip4p or tip5p
- - -[no]inter bool no Set the next 8 options to interactive
- - -[no]ss bool no Interactive SS bridge selection
- - -[no]ter bool no Interactive termini selection, instead of charged
(default)
- - -[no]lys bool no Interactive lysine selection, instead of charged
- - -[no]arg bool no Interactive arginine selection, instead of charged
- - -[no]asp bool no Interactive aspartic acid selection, instead of
charged
- - -[no]glu bool no Interactive glutamic acid selection, instead of
charged
- - -[no]gln bool no Interactive glutamine selection, instead of
neutral
- - -[no]his bool no Interactive histidine selection, instead of
checking H-bonds
- - -angle real 135 Minimum hydrogen-donor-acceptor angle for a
H-bond (degrees)
- - -dist real 0.3 Maximum donor-acceptor distance for a H-bond (nm)
- - -[no]una bool no Select aromatic rings with united CH atoms on
phenylalanine, tryptophane and tyrosine
- - -[no]ignh bool no Ignore hydrogen atoms that are in the coordinate
file
- - -[no]missing bool no Continue when atoms are missing, dangerous
- - -[no]v bool no Be slightly more verbose in messages
- - -posrefc real 1000 Force constant for position restraints
- - -vsite enum none Convert atoms to virtual sites: none, hydrogens
or aromatics
- - -[no]heavyh bool no Make hydrogen atoms heavy
- - -[no]deuterate bool no Change the mass of hydrogens to 2 amu
- - -[no]chargegrp bool yes Use charge groups in the .rtp file
- - -[no]cmap bool yes Use cmap torsions (if enabled in the .rtp file)
- - -[no]renum bool no Renumber the residues consecutively in the output
- - -[no]rtpres bool no Use .rtp entry names as residue names
Select the Force Field:
- - From '/usr/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (with CMAP) - version 2.0
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
16: [DEPRECATED] Encad all-atom force field, using full solvent charges
17: [DEPRECATED] Encad all-atom force field, using scaled-down vacuum charges
18: [DEPRECATED] Gromacs force field (see manual)
19: [DEPRECATED] Gromacs force field with hydrogens for NMR
1
Using the Amber03 force field in directory amber03.ff
Opening force field file /usr/share/gromacs/top/amber03.ff/watermodels.dat
Select the Water Model:
1: TIP3P TIP 3-point, recommended
2: TIP4P TIP 4-point
3: TIP4P-Ew TIP 4-point optimized with Ewald
4: SPC simple point charge
5: SPC/E extended simple point charge
6: None
1
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.r2b
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.r2b
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.r2b
Reading aceala1.pdb...
Read 16 atoms
Analyzing pdb file
Splitting chemical chains based on TER records or chain id changing.
There are 1 chains and 0 blocks of water and 2 residues with 16 atoms
chain #res #atoms
1 ' ' 2 16
All occupancies are one
Opening force field file /usr/share/gromacs/top/amber03.ff/atomtypes.atp
Atomtype 1
Reading residue database... (amber03)
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.rtp
Residue 93
Sorting it all out...
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.rtp
Residue 109
Sorting it all out...
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.rtp
Residue 125
Sorting it all out...
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.hdb
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.hdb
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.hdb
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.n.tdb
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.c.tdb
Processing chain 1 (16 atoms, 2 residues)
Identified residue ACE1 as a starting terminus.
Identified residue ALA2 as a ending terminus.
8 out of 8 lines of specbond.dat converted successfully
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.arn
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.arn
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.arn
Checking for duplicate atoms....
Generating any missing hydrogen atoms and/or adding termini.
Now there are 2 residues with 17 atoms
Making bonds...
Number of bonds was 16, now 16
Generating angles, dihedrals and pairs...
Before cleaning: 31 pairs
Before cleaning: 31 dihedrals
Keeping all generated dihedrals
Making cmap torsions...There are 31 dihedrals, 3 impropers, 27 angles
31 pairs, 16 bonds and 0 virtual sites
Total mass 130.124 a.m.u.
Total charge -1.000 e
Writing topology
Writing coordinate file...
--------- PLEASE NOTE ------------
You have successfully generated a topology from: aceala1.pdb.
The Amber03 force field and the tip3p water model are used.
--------- ETON ESAELP ------------
gcq#76: "It Costs Too Much If It Costs a Lot" (Magnapop)
Now create em.mdp. It's a small file with important configurations. I won't get into the meaning of each line.
integrator = steep nsteps = 25 pbc = no emtol = 100
grompp -c aceala1.gro -f em.mdp
:-) G R O M A C S (-:
Good gRace! Old Maple Actually Chews Slate
:-) VERSION 4.6.5 (-:
Contributions from Mark Abraham, Emile Apol, Rossen Apostolov,
Herman J.C. Berendsen, Aldert van Buuren, Pär Bjelkmar,
Rudi van Drunen, Anton Feenstra, Gerrit Groenhof, Christoph Junghans,
Peter Kasson, Carsten Kutzner, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2012,2013, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.
:-) grompp (-:
Option Filename Type Description
- - ------------------------------------------------------------
-f em.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c aceala1.gmx.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p topol.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o topol.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
- - -ref rotref.trr In/Out, Opt. Full precision trajectory: trr trj cpt
Option Type Value Description
- - ------------------------------------------------------
- - -[no]h bool no Print help info and quit
- - -[no]version bool no Print version info and quit
- - -nice int 0 Set the nicelevel
- - -[no]v bool no Be loud and noisy
- - -time real -1 Take frame at or first after this time.
- - -[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
- - -maxwarn int 0 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
- - -[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
- - -[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.1#
Generated 2278 of the 2278 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 2278 of the 2278 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'Protein'
NOTE 1 [file topol.top, line 194]:
System has non-zero total charge: -1.000000
Total charge should normally be an integer. See
http://www.gromacs.org/Documentation/Floating_Point_Arithmetic
for discussion on how close it should be to an integer.
Analysing residue names:
There are: 2 Protein residues
Analysing Protein...
Number of degrees of freedom in T-Coupling group rest is 48.00
NOTE 2 [file em.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 0 Mb of data
There were 2 notes
gcq#11: "She's Not Bad, She's Just Genetically Mean" (Captain Beefheart)
Done. Notice that there are two notes. We'll ignore them. Now run the minimization.
mdrun -v
:-) G R O M A C S (-:
Grunge ROck MAChoS
:-) VERSION 4.6.5 (-:
Contributions from Mark Abraham, Emile Apol, Rossen Apostolov,
Herman J.C. Berendsen, Aldert van Buuren, Pär Bjelkmar,
Rudi van Drunen, Anton Feenstra, Gerrit Groenhof, Christoph Junghans,
Peter Kasson, Carsten Kutzner, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2012,2013, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU Lesser General Public License
as published by the Free Software Foundation; either version 2.1
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
- - ------------------------------------------------------------
-s topol.tpr Input Run input file: tpr tpb tpa
-o traj.trr Output Full precision trajectory: trr trj cpt
-x traj.xtc Output, Opt. Compressed trajectory (portable xdr format)
- - -cpi state.cpt Input, Opt. Checkpoint file
- - -cpo state.cpt Output, Opt. Checkpoint file
-c confout.gro Output Structure file: gro g96 pdb etc.
-e ener.edr Output Energy file
-g md.log Output Log file
- - -dhdl dhdl.xvg Output, Opt. xvgr/xmgr file
- - -field field.xvg Output, Opt. xvgr/xmgr file
- - -table table.xvg Input, Opt. xvgr/xmgr file
- - -tabletf tabletf.xvg Input, Opt. xvgr/xmgr file
- - -tablep tablep.xvg Input, Opt. xvgr/xmgr file
- - -tableb table.xvg Input, Opt. xvgr/xmgr file
- - -rerun rerun.xtc Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
- - -tpi tpi.xvg Output, Opt. xvgr/xmgr file
- - -tpid tpidist.xvg Output, Opt. xvgr/xmgr file
-ei sam.edi Input, Opt. ED sampling input
-eo edsam.xvg Output, Opt. xvgr/xmgr file
-j wham.gct Input, Opt. General coupling stuff
-jo bam.gct Output, Opt. General coupling stuff
- - -ffout gct.xvg Output, Opt. xvgr/xmgr file
- - -devout deviatie.xvg Output, Opt. xvgr/xmgr file
- - -runav runaver.xvg Output, Opt. xvgr/xmgr file
-px pullx.xvg Output, Opt. xvgr/xmgr file
-pf pullf.xvg Output, Opt. xvgr/xmgr file
-ro rotation.xvg Output, Opt. xvgr/xmgr file
-ra rotangles.log Output, Opt. Log file
-rs rotslabs.log Output, Opt. Log file
-rt rottorque.log Output, Opt. Log file
- - -mtx nm.mtx Output, Opt. Hessian matrix
-dn dipole.ndx Output, Opt. Index file
- - -multidir rundir Input, Opt., Mult. Run directory
- - -membed membed.dat Input, Opt. Generic data file
-mp membed.top Input, Opt. Topology file
-mn membed.ndx Input, Opt. Index file
Option Type Value Description
- - ------------------------------------------------------
- - -[no]h bool no Print help info and quit
- - -[no]version bool no Print version info and quit
- - -nice int 0 Set the nicelevel
- - -deffnm string Set the default filename for all file options
- - -xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
- - -[no]pd bool no Use particle decompostion
- - -dd vector 0 0 0 Domain decomposition grid, 0 is optimize
- - -ddorder enum interleave DD node order: interleave, pp_pme or cartesian
- - -npme int -1 Number of separate nodes to be used for PME, -1
is guess
- - -nt int 0 Total number of threads to start (0 is guess)
- - -ntmpi int 0 Number of thread-MPI threads to start (0 is guess)
- - -ntomp int 0 Number of OpenMP threads per MPI process/thread
to start (0 is guess)
- - -ntomp_pme int 0 Number of OpenMP threads per MPI process/thread
to start (0 is -ntomp)
- - -pin enum auto Fix threads (or processes) to specific cores:
auto, on or off
- - -pinoffset int 0 The starting logical core number for pinning to
cores; used to avoid pinning threads from
different mdrun instances to the same core
- - -pinstride int 0 Pinning distance in logical cores for threads,
use 0 to minimize the number of threads per
physical core
- - -gpu_id string List of GPU device id-s to use, specifies the
per-node PP rank to GPU mapping
- - -[no]ddcheck bool yes Check for all bonded interactions with DD
- - -rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
- - -rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
- - -dlb enum auto Dynamic load balancing (with DD): auto, no or yes
- - -dds real 0.8 Minimum allowed dlb scaling of the DD cell size
- - -gcom int -1 Global communication frequency
- - -nb enum auto Calculate non-bonded interactions on: auto, cpu,
gpu or gpu_cpu
- - -[no]tunepme bool yes Optimize PME load between PP/PME nodes or GPU/CPU
- - -[no]testverlet bool no Test the Verlet non-bonded scheme
- - -[no]v bool yes Be loud and noisy
- - -[no]compact bool yes Write a compact log file
- - -[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
- - -pforce real -1 Print all forces larger than this (kJ/mol nm)
- - -[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
- - -cpt real 15 Checkpoint interval (minutes)
- - -[no]cpnum bool no Keep and number checkpoint files
- - -[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
- - -nsteps step -2 Run this number of steps, overrides .mdp file
option
- - -maxh real -1 Terminate after 0.99 times this time (hours)
- - -multi int 0 Do multiple simulations in parallel
- - -replex int 0 Attempt replica exchange periodically with this
period (steps)
- - -nex int 0 Number of random exchanges to carry out each
exchange interval (N^3 is one suggestion). -nex
zero or not specified gives neighbor replica
exchange.
- - -reseed int -1 Seed for replica exchange, -1 is generate a seed
- - -[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Reading file topol.tpr, VERSION 4.6.5 (single precision)
NOTE: Parallelization is limited by the small number of atoms,
only starting 1 thread-MPI threads.
You can use the -nt and/or -ntmpi option to optimize the number of threads.
Using 1 MPI thread
Steepest Descents:
Tolerance (Fmax) = 1.00000e+02
Number of steps = 25
Step= 0, Dmax= 1.0e-02 nm, Epot= 3.37229e+02 Fmax= 1.41051e+04, atom= 16
Step= 1, Dmax= 1.0e-02 nm, Epot= 1.38289e+02 Fmax= 3.90973e+03, atom= 16
Step= 2, Dmax= 1.2e-02 nm, Epot= 6.82132e+01 Fmax= 4.33682e+03, atom= 15
Step= 4, Dmax= 7.2e-03 nm, Epot= 4.93446e+01 Fmax= 4.35624e+03, atom= 15
Step= 6, Dmax= 4.3e-03 nm, Epot= 3.40453e+01 Fmax= 1.83079e+03, atom= 15
Step= 8, Dmax= 2.6e-03 nm, Epot= 3.02945e+01 Fmax= 2.33007e+03, atom= 15
Step= 9, Dmax= 3.1e-03 nm, Epot= 2.73672e+01 Fmax= 2.23327e+03, atom= 15
Step= 11, Dmax= 1.9e-03 nm, Epot= 2.32383e+01 Fmax= 8.05830e+02, atom= 15
Step= 12, Dmax= 2.2e-03 nm, Epot= 2.08665e+01 Fmax= 2.11304e+03, atom= 15
Step= 13, Dmax= 2.7e-03 nm, Epot= 1.91569e+01 Fmax= 2.13703e+03, atom= 15
Step= 15, Dmax= 1.6e-03 nm, Epot= 1.62574e+01 Fmax= 5.19615e+02, atom= 85
Step= 16, Dmax= 1.9e-03 nm, Epot= 1.39709e+01 Fmax= 1.62357e+03, atom= 15
Step= 17, Dmax= 2.3e-03 nm, Epot= 1.34916e+01 Fmax= 1.91558e+03, atom= 15
Step= 19, Dmax= 1.4e-03 nm, Epot= 1.11926e+01 Fmax= 3.85227e+02, atom= 85
Step= 20, Dmax= 1.7e-03 nm, Epot= 9.33456e+00 Fmax= 1.32394e+03, atom= 15
Step= 21, Dmax= 2.0e-03 nm, Epot= 9.21982e+00 Fmax= 1.74297e+03, atom= 15
Step= 22, Dmax= 2.4e-03 nm, Epot= 8.95828e+00 Fmax= 1.98853e+03, atom= 15
Step= 24, Dmax= 1.4e-03 nm, Epot= 6.72025e+00 Fmax= 2.99685e+02, atom= 85
Step= 25, Dmax= 1.7e-03 nm, Epot= 5.88220e+00 Fmax= 1.87057e+03, atom= 15
Energy minimization reached the maximum number of steps before the forces
reached the requested precision Fmax < 100.
writing lowest energy coordinates.
Steepest Descents did not converge to Fmax < 100 in 26 steps.
Potential Energy = 5.8822021e+00
Maximum force = 1.8705703e+03 on atom 15
Norm of force = 5.7199799e+02
gcq#213: "Engage" (J.L. Picard)
At this point we can obviously see that we should have more steps. Potential energy went from 337 kJ/mol to 5 kJ/mol in 25 steps. Therefore, another 25 steps would be worthwhile. Let's try with 100 steps. em100.mdp now says steps = 100
Steps from 25 to 100: Step= 25, Dmax= 1.7e-03 nm, Epot= 5.88220e+00 Fmax= 1.87057e+03, atom= 15 Step= 26, Dmax= 2.1e-03 nm, Epot= 4.63776e+00 Fmax= 1.28671e+03, atom= 15 Step= 28, Dmax= 1.2e-03 nm, Epot= 3.64270e+00 Fmax= 6.75797e+02, atom= 15 Step= 30, Dmax= 7.5e-04 nm, Epot= 3.15042e+00 Fmax= 4.31048e+02, atom= 15 Step= 31, Dmax= 9.0e-04 nm, Epot= 2.84302e+00 Fmax= 1.03663e+03, atom= 15 Step= 32, Dmax= 1.1e-03 nm, Epot= 2.18570e+00 Fmax= 5.76095e+02, atom= 15 Step= 34, Dmax= 6.5e-04 nm, Epot= 1.74603e+00 Fmax= 4.73068e+02, atom= 15 Step= 35, Dmax= 7.8e-04 nm, Epot= 1.36945e+00 Fmax= 6.40599e+02, atom= 15 Step= 36, Dmax= 9.3e-04 nm, Epot= 1.05707e+00 Fmax= 8.62854e+02, atom= 15 Step= 37, Dmax= 1.1e-03 nm, Epot= 6.80450e-01 Fmax= 7.92532e+02, atom= 15 Step= 39, Dmax= 6.7e-04 nm, Epot= 1.37573e-01 Fmax= 2.85551e+02, atom= 15 Step= 40, Dmax= 8.0e-04 nm, Epot= -3.20038e-01 Fmax= 7.97426e+02, atom= 15 Step= 41, Dmax= 9.7e-04 nm, Epot= -7.21436e-01 Fmax= 7.51710e+02, atom= 15 Step= 42, Dmax= 1.2e-03 nm, Epot= -8.87543e-01 Fmax= 9.49344e+02, atom= 15 Step= 43, Dmax= 1.4e-03 nm, Epot= -9.74335e-01 Fmax= 1.26718e+03, atom= 15 Step= 44, Dmax= 1.7e-03 nm, Epot= -1.25046e+00 Fmax= 1.22974e+03, atom= 15 Step= 46, Dmax= 1.0e-03 nm, Epot= -2.18298e+00 Fmax= 3.56542e+02, atom= 15 Step= 48, Dmax= 6.0e-04 nm, Epot= -2.49353e+00 Fmax= 4.74792e+02, atom= 15 Step= 49, Dmax= 7.2e-04 nm, Epot= -2.73279e+00 Fmax= 7.15007e+02, atom= 15 Step= 50, Dmax= 8.7e-04 nm, Epot= -3.05582e+00 Fmax= 5.50525e+02, atom= 15 Step= 52, Dmax= 5.2e-04 nm, Epot= -3.39255e+00 Fmax= 3.01295e+02, atom= 15 Step= 53, Dmax= 6.2e-04 nm, Epot= -3.69717e+00 Fmax= 5.39593e+02, atom= 15 Step= 54, Dmax= 7.5e-04 nm, Epot= -3.93311e+00 Fmax= 6.85335e+02, atom= 15 Step= 55, Dmax= 9.0e-04 nm, Epot= -4.18774e+00 Fmax= 6.22661e+02, atom= 15 Step= 57, Dmax= 5.4e-04 nm, Epot= -4.54706e+00 Fmax= 2.55038e+02, atom= 15 Step= 58, Dmax= 6.5e-04 nm, Epot= -4.85925e+00 Fmax= 5.91890e+02, atom= 15 Step= 59, Dmax= 7.8e-04 nm, Epot= -5.09546e+00 Fmax= 6.71699e+02, atom= 15 Step= 60, Dmax= 9.3e-04 nm, Epot= -5.29047e+00 Fmax= 6.82407e+02, atom= 15 Step= 62, Dmax= 5.6e-04 nm, Epot= -5.67368e+00 Fmax= 2.23499e+02, atom= 15 Step= 63, Dmax= 6.7e-04 nm, Epot= -5.99097e+00 Fmax= 6.31567e+02, atom= 15 Step= 64, Dmax= 8.0e-04 nm, Epot= -6.22238e+00 Fmax= 6.73722e+02, atom= 15 Step= 65, Dmax= 9.6e-04 nm, Epot= -6.36801e+00 Fmax= 7.30559e+02, atom= 15 Step= 67, Dmax= 5.8e-04 nm, Epot= -6.77222e+00 Fmax= 2.05365e+02, atom= 15 Step= 68, Dmax= 6.9e-04 nm, Epot= -7.07648e+00 Fmax= 6.65278e+02, atom= 15 Step= 69, Dmax= 8.3e-04 nm, Epot= -7.30093e+00 Fmax= 6.83842e+02, atom= 15 Step= 70, Dmax= 1.0e-03 nm, Epot= -7.40231e+00 Fmax= 7.73894e+02, atom= 15 Step= 72, Dmax= 6.0e-04 nm, Epot= -7.82770e+00 Fmax= 1.93378e+02, atom= 15 Step= 73, Dmax= 7.2e-04 nm, Epot= -8.10611e+00 Fmax= 6.98348e+02, atom= 15 Step= 74, Dmax= 8.6e-04 nm, Epot= -8.32544e+00 Fmax= 6.96258e+02, atom= 15 Step= 75, Dmax= 1.0e-03 nm, Epot= -8.38281e+00 Fmax= 8.17505e+02, atom= 15 Step= 77, Dmax= 6.2e-04 nm, Epot= -8.83289e+00 Fmax= 1.82288e+02, atom= 15 Step= 78, Dmax= 7.5e-04 nm, Epot= -9.08395e+00 Fmax= 7.31604e+02, atom= 15 Step= 79, Dmax= 9.0e-04 nm, Epot= -9.30014e+00 Fmax= 7.10133e+02, atom= 15 Step= 80, Dmax= 1.1e-03 nm, Epot= -9.31290e+00 Fmax= 8.62212e+02, atom= 15 Step= 82, Dmax= 6.4e-04 nm, Epot= -9.79193e+00 Fmax= 1.71301e+02, atom= 15 Step= 83, Dmax= 7.7e-04 nm, Epot= -1.00156e+01 Fmax= 7.64914e+02, atom= 15 Step= 84, Dmax= 9.3e-04 nm, Epot= -1.02304e+01 Fmax= 7.25622e+02, atom= 15 Step= 86, Dmax= 5.6e-04 nm, Epot= -1.05448e+01 Fmax= 1.69183e+02, atom= 17 Step= 87, Dmax= 6.7e-04 nm, Epot= -1.07820e+01 Fmax= 6.67437e+02, atom= 15 Step= 88, Dmax= 8.0e-04 nm, Epot= -1.09632e+01 Fmax= 5.05086e+02, atom= 15 Step= 90, Dmax= 4.8e-04 nm, Epot= -1.11735e+01 Fmax= 2.84477e+02, atom= 15 Step= 91, Dmax= 5.8e-04 nm, Epot= -1.12706e+01 Fmax= 4.98466e+02, atom= 15 Step= 92, Dmax= 6.9e-04 nm, Epot= -1.13672e+01 Fmax= 6.36931e+02, atom= 15 Step= 93, Dmax= 8.3e-04 nm, Epot= -1.14917e+01 Fmax= 5.76544e+02, atom= 15 Step= 95, Dmax= 5.0e-04 nm, Epot= -1.17386e+01 Fmax= 2.35424e+02, atom= 15 Step= 96, Dmax= 6.0e-04 nm, Epot= -1.18294e+01 Fmax= 5.54518e+02, atom= 15 Step= 97, Dmax= 7.2e-04 nm, Epot= -1.19516e+01 Fmax= 6.14315e+02, atom= 15 Step= 98, Dmax= 8.6e-04 nm, Epot= -1.20219e+01 Fmax= 6.42397e+02, atom= 15 Step= 100, Dmax= 5.2e-04 nm, Epot= -1.23065e+01 Fmax= 1.94291e+02, atom= 15Now the potential energy is at -12.3 kJ/mol, which is quite good. It didn't converge because it's still on its way to perfect. Let's take a look at it.
editconf -f confout.gro -o aceala1_min.pdbNow open it in PyMOL.
pymol load aceala1_min.pdbThe following commands will make it look like a ball and stick model:
hide everything show sticks show spheres set stick_radius, .07 set sphere_scale, .18 set sphere_scale, .13, elem H set bg_rgb=[1, 1, 1] set stick_quality, 50 set sphere_quality, 4 color gray85, elem C color red, elem O color slate, elem N color gray98, elem H set stick_color, black set ray_trace_mode, 1 set ray_texture, 2 set antialias, 3 set ambient, 0.5 set spec_count, 5 set shininess, 50 set specular, 1 set reflect, .1To get a nice rendering, run:
ray
PyMOL is a very complex program which can do an impressive list of things. I won't get into exactly what you can do with it, but believe me it's extensive. The protein creation we did at the beginning was just one piece of it's code.
It turns out that the minimization converges at 166 steps, so it's not too far away from done. The potential energy it settles down into is -17.5 kJ/mol, only 5 more units than what we had. Surprising though is that the difference in the two models is very small.
If you want to look at the trajectory, try this:
trjconv -f traj.trr -o aceala1_trj.pdb
It will ask for the group. Group 0 is everything, so pick it.
The next step in our journey is to scale up our operations. Instead of making a two-item protein, let's minimize a real protein.
RCSB PDB contains a list of proteins that have been studied. It's easy to look through them. Of course there are a lot, so downloading them all should be done via http://www.wwpdb.org/downloads.html
I picked a random one, 1OEI and downloaded the pdb. You're welcome to look for a different protein but many proteins use modified residues, so they will cause GROMACS to throw errors. For example, I wanted to minimize 3RJV because it is currently being studied by Fold.it. It turns out that it uses a modified residue, MSE. MSE contains selenium, which is a fairly interesting semiconductor. MSE can be found in Brazil nuts, cereal grains, soybeans, and grassland legumes according to Wikipedia.
So let's minimize our real protein.
Since GROMACS has many default files that we'll be using, it makes sense to create a directory and work in it for the duration of your work. I use just one non-default file and the reason for that is just habit.
If you run this: pdb2gmx -f 1OEI.pdb -o 1OEI.gro
you will get an error complaining about:
"Atom HB3 in residue HIS 61 was not found in rtp entry NHID with 19 atoms
while sorting atoms."
This is because the naming of atoms in residues is not completely standardized. Therefore hydrogens are often incorrectly named in PDB files. GROMACS has a flag to solve that called "-ignh". It ignores hydrogens when trying to figure out what is connected to what. Since hydrogens can only have one bond, they are always only connected to one thing, so they can be safely ignored most of the time.
pdb2gmx -f 1OEI.pdb -o 1OEI.gro -ignh
:-) G R O M A C S (-:
Great Red Owns Many ACres of Sand
:-) VERSION 4.6.5 (-:
:-) pdb2gmx (-:
Select the Force Field:
- - From '/usr/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (with CMAP) - version 2.0
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
16: [DEPRECATED] Encad all-atom force field, using full solvent charges
17: [DEPRECATED] Encad all-atom force field, using scaled-down vacuum charges
18: [DEPRECATED] Gromacs force field (see manual)
19: [DEPRECATED] Gromacs force field with hydrogens for NMR
1
Using the Amber03 force field in directory amber03.ff
Opening force field file /usr/share/gromacs/top/amber03.ff/watermodels.dat
Select the Water Model:
1: TIP3P TIP 3-point, recommended
2: TIP4P TIP 4-point
3: TIP4P-Ew TIP 4-point optimized with Ewald
4: SPC simple point charge
5: SPC/E extended simple point charge
6: None
1
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.r2b
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.r2b
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.r2b
Reading 1OEI.pdb...
Read 'MAJOR PRION PROTEIN', 168 atoms
Analyzing pdb file
Splitting chemical chains based on TER records or chain id changing.
There are 1 chains and 0 blocks of water and 84 residues with 168 atoms
chain #res #atoms
1 'A' 24 168
All occupancies are one
Opening force field file /usr/share/gromacs/top/amber03.ff/atomtypes.atp
Atomtype 1
Reading residue database... (amber03)
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.rtp
Residue 93
Sorting it all out...
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.rtp
Residue 109
Sorting it all out...
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.rtp
Residue 125
Sorting it all out...
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.hdb
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.hdb
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.hdb
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.n.tdb
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.c.tdb
Back Off! I just backed up topol.top to ./#topol.top.1#
Processing chain 1 'A' (168 atoms, 24 residues)
Analysing hydrogen-bonding network for automated assignment of histidine
protonation. 36 donors and 27 acceptors were found.
There are 39 hydrogen bonds
Will use HISD for residue 61
Will use HISE for residue 69
Will use HISE for residue 77
Identified residue HIS61 as a starting terminus.
Identified residue PRO84 as a ending terminus.
8 out of 8 lines of specbond.dat converted successfully
Special Atom Distance matrix:
HIS61 HIS69
NE210 NE266
HIS69 NE266 1.244
HIS77 NE2122 1.451 1.232
Opening force field file /usr/share/gromacs/top/amber03.ff/aminoacids.arn
Opening force field file /usr/share/gromacs/top/amber03.ff/dna.arn
Opening force field file /usr/share/gromacs/top/amber03.ff/rna.arn
Checking for duplicate atoms....
Generating any missing hydrogen atoms and/or adding termini.
Now there are 24 residues with 303 atoms
Making bonds...
Number of bonds was 315, now 314
Generating angles, dihedrals and pairs...
Before cleaning: 779 pairs
Before cleaning: 824 dihedrals
Keeping all generated dihedrals
Making cmap torsions...There are 824 dihedrals, 86 impropers, 555 angles
770 pairs, 314 bonds and 0 virtual sites
Total mass 2348.482 a.m.u.
Total charge -0.000 e
Writing topology
Writing coordinate file...
--------- PLEASE NOTE ------------
You have successfully generated a topology from: 1OEI.pdb.
The Amber03 force field and the tip3p water model are used.
--------- ETON ESAELP ------------
gcq#285: "Shaken, not Stirred" (J. Bond)
We modify our em.mdp to use 10,000 step instead of 100, naming it em10k.mdp. We use this file to setup grompp to create a correct mdout.mdp and topol.tpr.
cp -i ../test1/em10k.mdp .
grompp -c 1OEI.gro -f em10k.mdp
:-) G R O M A C S (-:
GROwing Monsters And Cloning Shrimps
:-) VERSION 4.6.5 (-:
:-) grompp (-:
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.2#
Generated 2278 of the 2278 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 2278 of the 2278 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'Protein_chain_A'
Analysing residue names:
There are: 24 Protein residues
Analysing Protein...
Number of degrees of freedom in T-Coupling group rest is 906.00
NOTE 1 [file em10k.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 0 Mb of data
There was 1 note
Back Off! I just backed up topol.tpr to ./#topol.tpr.1#
gcq#123: "I'm Gonna Get Medieval On Your Ass" (Pulp Fiction)
Now we're ready to minimize.
time mdrun -v
:-) G R O M A C S (-:
Glycine aRginine prOline Methionine Alanine Cystine Serine
:-) VERSION 4.6.5 (-:
:-) mdrun (-:
Back Off! I just backed up md.log to ./#md.log.1#
Reading file topol.tpr, VERSION 4.6.5 (single precision)
NOTE: Parallelization is limited by the small number of atoms,
only starting 3 thread-MPI threads.
You can use the -nt and/or -ntmpi option to optimize the number of threads.
Using 3 MPI threads
NOTE: The number of threads is not equal to the number of (logical) cores
and the -pin option is set to auto: will not pin thread to cores.
This can lead to significant performance degradation.
Consider using -pin on (and -pinoffset in case you run multiple jobs).
Back Off! I just backed up traj.trr to ./#traj.trr.1#
Back Off! I just backed up ener.edr to ./#ener.edr.1#
Steepest Descents:
Tolerance (Fmax) = 1.00000e+02
Number of steps = 10000
Step= 0, Dmax= 1.0e-02 nm, Epot= 1.10861e+03 Fmax= 1.39522e+04, atom= 302
Step= 1, Dmax= 1.0e-02 nm, Epot= 2.98488e+02 Fmax= 3.68642e+03, atom= 303
Step= 2, Dmax= 1.2e-02 nm, Epot= -7.94045e+01 Fmax= 5.03219e+03, atom= 301
Step= 4, Dmax= 7.2e-03 nm, Epot= -3.19202e+02 Fmax= 3.97506e+03, atom= 301
Step= 7, Dmax= 2.2e-03 nm, Epot= -3.33482e+02 Fmax= 9.49081e+02, atom= 301
Step= 8, Dmax= 2.6e-03 nm, Epot= -3.67506e+02 Fmax= 2.36492e+03, atom= 301
Step= 9, Dmax= 3.1e-03 nm, Epot= -4.09318e+02 Fmax= 1.95051e+03, atom= 301
Step= 16, Dmax= 5.8e-05 nm, Epot= -4.09757e+02 Fmax= 1.86791e+03, atom= 301
Step= 21, Dmax= 4.4e-06 nm, Epot= -4.09789e+02 Fmax= 1.86168e+03, atom= 301
Step= 24, Dmax= 1.3e-06 nm, Epot= -4.09801e+02 Fmax= 1.85984e+03, atom= 301
Step= 25, Dmax= 1.6e-06 nm, Epot= -3.86413e+02 Fmax= 1.83612e+03, atom= 301
Energy minimization has stopped, but the forces have not converged to the
requested precision Fmax < 100 (which may not be possible for your system).
It stopped because the algorithm tried to make a new step whose size was too
small, or there was no change in the energy since last step. Either way, we
regard the minimization as converged to within the available machine
precision, given your starting configuration and EM parameters.
Double precision normally gives you higher accuracy, but this is often not
needed for preparing to run molecular dynamics.
writing lowest energy coordinates.
Back Off! I just backed up confout.gro to ./#confout.gro.1#
Steepest Descents converged to machine precision in 26 steps,
but did not reach the requested Fmax < 100.
Potential Energy = -4.0980066e+02
Maximum force = 1.8598427e+03 on atom 301
Norm of force = 2.1627922e+02
gcq#308: "California, R.I.P." (Red Hot Chili Peppars)
real 0m0.059s
user 0m0.100s
sys 0m0.048s
It finished very quickly in just 25 steps. It ended up with potential energy of -386 kJ/mol. Of course this file has already been minimized. If you look at the input file in PyMOL, you will notice that there are 20 models, GROMACS only worked on the first. If you're interested, try minimizing the others. You might have noticed the warning in the output "Energy minimization has stopped, but the forces have not converged to the requested precision Fmax < 100 (which may not be possible for your system)." This means that the forces are higher than we want them to be. In this case, they're 1836 kJ mol-1 nm-1. This is a lot higher than we intended, but that's okay since we successfully minimized it.
Let's visualize the output.
editconf -f confout.gro -o 1OEI_min.pdb
:-) G R O M A C S (-:
GROtesk MACabre and Sinister
:-) VERSION 4.6.5 (-:
:-) editconf (-:
Read 303 atoms
Volume: 0.001 nm^3, corresponds to roughly 0 electrons
No velocities found
gcq#107: "Ease Myself Into the Body Bag" (P.J. Harvey)
Below you can see my rendering as a cartoon. Of course proteins can be rendered in multiple ways, ball and stick gives you a slightly more accurate rendering while the cartoon gives you an overview of the structure.

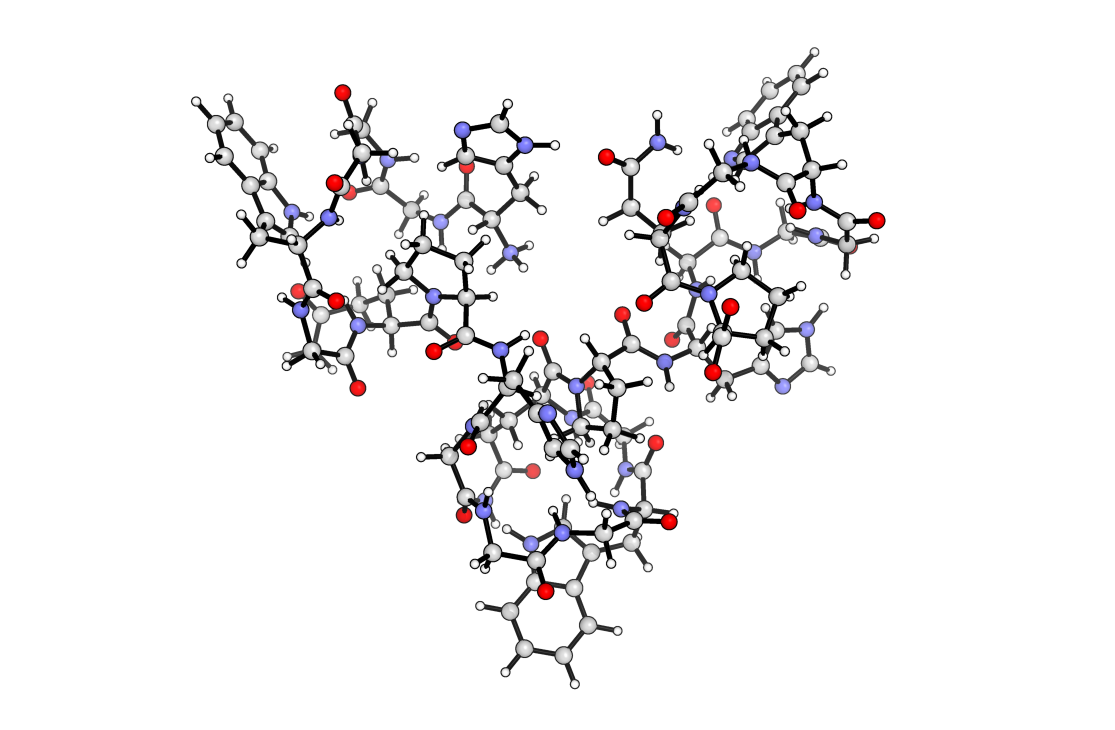
This concludes the tutorial. In this tutorial we
- Created a peptide with PyMOL.
- Minimized it with GROMACS.
- Downloaded a real protein.
- Minimized it with GROMACS.
Most people ask how it's going. It's difficult and fun. I expected it to be difficult and I've spent a lot of time in the past 3.5 months doing things that are not directly related to nanotechnology. But I've had fun doing nanotech and I've had fun doing other stuff.
I attended a seminar series at the University of Washington's Molecular Engineering and Sciences Institute. The next one starts at the end of September if you're interested in attending.
I finished reading Nanosystems and compiled a list of things to ask Dr. K. Eric Drexler. A full book report will follow in the future.
Before I go I'd like to invite hackers amongst my readers to come to Neg9 Seattle Summer Hack Fest on July 3, 2014. It will not focus on nanotech nor talking, just hacking. If I don't write before Friday, have a wonderful Independence Day. This year I'm celebrating independence from a 9-to-5 job.
Javantea out.
-----BEGIN PGP SIGNATURE----- Version: GnuPG v2.0.22 (GNU/Linux) iEYEARECAAYFAlOy3WAACgkQpmRrABlU/tJg9wCdH1S+2lR6EALbNZp9STDRVy+f Q0oAoJPMmP979l5Qt7GOpTT8flUu6pvC =/vRP -----END PGP SIGNATURE-----Permalink
-
Leave a Reply




Comments: 0
Leave a reply »